Compreendendo as malformações cardíacas congênitas
A palavra "congênita" significa algo que existe ao nascimento. Um defeito cardíaco congênito (CHD) é um problema estrutural do coração que está presente no nascimento e ocorre quando o coração, ou vasos sanguíneos próximos ao coração, não se desenvolvem normalmente antes do nascimento.
Tais defeitos resultam quando ocorre uma alteração durante o desenvolvimento do coração logo após a concepção - muitas vezes antes que a mãe esteja ciente de que está grávida.
Tais problemas podem ou não ter um efeito perturbador no sistema circulatório de uma pessoa. Mas ter um defeito cardíaco congênito pode aumentar o risco de desenvolver certas condições médicas.
Para entender defeitos cardíacos congênitos, é útil lembrar como o coração deve funcionar:
Um coração normal tem válvulas, artérias e câmaras que circulam o sangue em um padrão recorrente: Quando todas as câmaras e válvulas funcionam corretamente, o sangue é bombeado através do coração, para os pulmões, de volta para o coração e, em seguida, por todo o corpo para entregar oxigênio. Quando as válvulas, câmaras, artérias e veias são malformadas, esse padrão de circulação pode ser prejudicado.
Os defeitos cardíacos congênitos variam em gravidade, desde problemas simples, como comunicações entre as câmaras do coração, até malformações maiores, como a completa ausência de uma ou mais câmaras ou válvulas.
Condições associadas
Um defeito cardíaco congênito pode aumentar o risco de certas condições médicas, incluindo:
- Hipertensão pulmonar
- Arritmias
- Endocardite infecciosa
- Alterações da coagulação
- Insuficiência cardíaca congestiva
Perguntas frequentes
Todos os problemas cardíacos em crianças são congênitos?
Não, mas a maioria é. Existem três categorias gerais de possíveis problemas cardíacos na infância: defeitos estruturais, danos adquiridos e distúrbios do ritmo cardíaco. Esses defeitos são geralmente, mas nem sempre, diagnosticados no início da vida.
As crianças também podem nascer ou desenvolver problemas de frequência cardíaca, como batimentos cardíacos lentos, rápidos ou irregulares, conhecidos como “ arritmias ”.
Quem corre o risco de ter um filho com um defeito cardíaco congênito?
Qualquer um pode ter um filho com um defeito cardíaco congênito. Dos 1.000 nascimentos, pelo menos oito bebês terão algum tipo de distúrbio cardíaco congênito, a maioria dos quais é leve. Se você ou outros membros da família já tiveram um bebê com um problema cardíaco, o risco de ter um bebê com defeito cardíaco pode ser maior.
Por que defeitos cardíacos congênitos ocorrem?
Na maioria das vezes, a causa não é conhecida. Embora a razão pela qual os defeitos ocorrem seja supostamente genética, apenas alguns genes foram descobertos e ligados a defeitos cardíacos.
Raramente, a ingestão de alguns medicamentos e a ocorrência de algumas infecções durante a gravidez podem causar defeitos.
Como posso saber se meu bebê ou criança tem um defeito cardíaco congênito?
Os distúrbios cardíacos graves geralmente se tornam evidentes durante os primeiros meses após o nascimento. Alguns bebês têm cianose (alteração da cor relacionada com a baixa oxigenação) ou têm pressão arterial muito baixa logo após o nascimento. Outros defeitos causam dificuldades respiratórias, problemas de alimentação ou ganho de peso insuficiente.
Pequenos defeitos são mais frequentemente diagnosticados durante um exame médico de rotina. Enquanto a maioria dos sopros cardíacos em crianças é normal, alguns podem ser devido a defeitos cardíacos.
Quão bem as pessoas com defeitos cardíacos congênitos podem viver?
Praticamente todas as crianças com defeitos simples sobrevivem até a idade adulta. Embora a capacidade de exercício possa ser limitada, a maioria das pessoas leva uma vida normal ou quase normal.
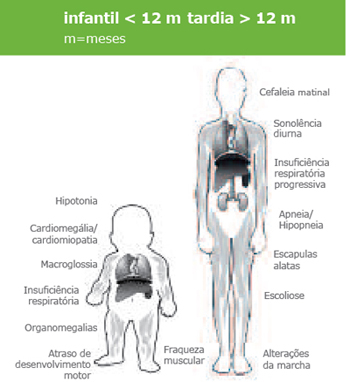
Com problemas mais complexos, as limitações são comuns. Algumas crianças com defeitos cardíacos congênitos têm atraso no desenvolvimento ou outras dificuldades de aprendizagem, especialmente quando estas cardiopatias são associadas a outras malformações (ou seja, quando são sindrômicas).
Qual a relação entre a genética e malformações cardíacas?
Acredita-se que a maioria dos defeitos cardíacos congênitos resulte de mutações genéticas. Isto foi sugerido pelas primeiras observações da herança evidente de cardiopatia em algumas famílias e identificação de maior risco de cardiopatia em bebês com irmãos acometidos. Outra evidência veio de síndromes congênitas devido a micro e macro deleções de regiões cromossômicas que resultariam em DAC juntamente com várias outras manifestações (apresentações chamadas sindrômicas). Nas últimas décadas, e com o advento do sequenciamento de genes e outras técnicas, tornou-se possível identificar muitas das causas genéticas das cardiopatias congênitas, mas nem todos os casos podem ser esclarecidos através das técnicas de investigação disponíveis atualmente.
Como é realizada a diferenciação entre cardiopatia congênita sindrômica e não sindrômica (ou isolada)?
Esta diferenciação pode ser realizada através do exame físico por um médico(a) geneticista ou por um profissional experiente na avaliação de defeitos congênitos. Além disso, exames complementares como ecografias ou radiografias podem ser necessários em casos específicos e conforme orientação clínica, buscando pela evidência de outras malformações associadas à cardiopatia.
Algumas das síndromes genéticas mais frequentemente associadas a cardiopatias são: Síndrome de Down, Síndrome de Turner, Síndrome Velo-cardio-facial ou DiGeorge, Síndrome de Charge e Síndrome de Noonan.
Cardiopatias congênitas: Classificação
| Categoria de classificação | Diagnósticos Mais Comuns |
| Anormalidades de posição e conexão do coração | Dextrocardia Ventrículo Esquerdo de Entrada Dupla
Situs inverso Atrial (DILV)
Transposição do Ventrículo Esquerdo com Dupla Entrada de Ventrículo Direito (DIRV)
Transposição de Grandes Artérias (TGA)
Ventrículo Direito de Saída Dupla (DORV)
Tronco Arterial Comum ou Truncus Arteriosus (TA) |
| Tetralogia de Fallot e variantes | Tetralogia de Fallot (TOF)
Atresia Pulmonar (PA) e Defeito Ventricular Septal (VSD) |
| Anormalidades de grandes veias | Anomalias de Veia Cava Superior (SVC) e
Veia Cava Inferior (SVC)
Anormalidades no Seio Coronariano
Conexão Anômala Venosa Pulmonar Total (CATVP)
Conexão Venosa Pulmonar Parcialmente Anômala (PAPVC) |
| Anormalidades dos átrios e septo atrial | Defeito septal atrial (ASD)
Forame oval patente (FOP) |
| Anormalidades das válvulas AV e defeito septal AV | Regurgitação tricúspide (TR)
Estenose tricúspide (TS)
Anomalia de Ebstein
Regurgitação mitral (RM)
Estenose mitral (EM)
Prolapso de Valvula Mitral(MVP)
Defeito do septo atrioventricular (AVSD) |
| Anormalidades dos ventrículos e do septo ventricular | Ventrículo Único
Desequilíbrio ventricular: VD dominante + VD hipoplásico, ou VD dominante + aneurisma hipoplásico do VD (VD, VE ou septal)
Síndrome do Coração Esquerdo Hipoplásico (SCEh)
Ventrículo Direito de Câmara Dividida (DDV)
Comunicação Interventricular
(CIV) |
| Anormalidades de válvulas AV e grandes artérias | Janela Aortopulmonar (Janela AP)
Estenose Pulmonar (PS), Valvar ou Subalvar
Estenose da Artéria Pulmonar (SAP)
Estenose Aórtica (EA)
Insuficiência aórtica (IA)
Válvula Aórtica Bicúspide (VAB)
Estenose Aórtica Supravalvar (SVS)
Coarctação da Aorta (COA)
Aortic Aortic Interrompida (IAA) |
| Anormalidades das artérias coronárias, ducto arterial e pericárdio; Fístula AV | Origem Anômala da Artéria Coronária da Artéria Pulmonar (ALCAPA)
Persistência do Ductus Arteriosus (PDA) |