No dia 5 de maio é lembrado o Dia da Síndrome de Cri du Chat.
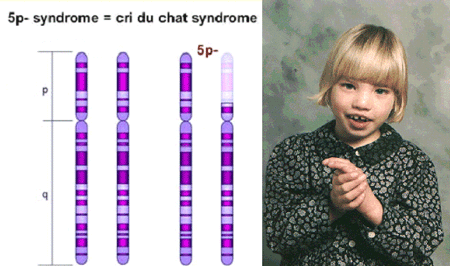
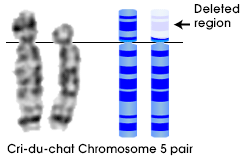
A Síndrome de Cri-du-chat, também conhecida como síndrome 5p- ("5 p menos"), é uma condição cromossômica que resulta quando uma parte do cromossomo 5 está faltando. Bebês com esta condição muitas vezes têm choro agudo, o que levou ao nome da síndrome na época de suas primeiras descrições.
O distúrbio é caracterizado por deficiência intelectual e atraso no desenvolvimento, tamanho pequeno da cabeça (microcefalia), baixo peso ao nascer e tônus muscular diminuído (hipotonia) na infância.
Indivíduos afetados também possuem características faciais distintas, incluindo olhos amplamente espaçados (hipertelorismo), orelhas de implantação baixa, uma mandíbula pequena e um rosto arredondado. Algumas crianças com síndrome de cri-du-chat podem apresentar malformações cardíacas.
A síndrome de Cri-du-chat ocorre em cerca de 1 em cada 20.000 a 50.000 recém-nascidos. Esta condição é encontrada em pessoas de todas as origens étnicas.
A maioria dos casos de síndrome de cri-du-chat não é herdada. A deleção ocorre mais frequentemente como um evento aleatório durante a formação de células reprodutivas (óvulos ou espermatozóides) ou no desenvolvimento fetal precoce. As pessoas afetadas geralmente não têm histórico desta condição em sua família. Para aconselhamento genético sempre procure seu médico(a) geneticista.

O diagnóstico desta condição é suspeitado após avaliação clínica com história médica e exame físico dismorfológico. Fazer um diagnóstico para uma doença genética ou rara muitas vezes pode ser um desafio. Os profissionais de saúde geralmente examinam o histórico médico, os sintomas, o exame físico e os resultados dos exames laboratoriais de uma pessoa para fazer um diagnóstico. Posteriormente pode ser solicitado um teste dirigido específico para essa condição, buscando avaliar a presença ou não da região que está deletada nesta síndrome. Se você tiver dúvidas sobre como obter um diagnóstico, entre em contato com um geneticista clínico.
Como não há até o momento tratamento específico disponível para a Síndrome de Cri du chat, a intervenção precoce é recomendada nas áreas de fisioterapia (atingir metas físicas e motoras como sentar e levantar), comunicação (fonoaudiologia, instrução em língua de sinais caso a caso), terapia comportamental (hiperatividade, falta de atenção, agressão) e aprendizagem (educação especial). Como os sintomas podem variar de indivíduo para indivíduo, recomenda-se discutir essas opções com um profissional de saúde para desenvolver um plano personalizado para terapia.